


Neuroinflammation in ME/CFS and Fibromyalgia
In previous articles I’ve been trying to set out, at some length and based on converging evidence, a proposed model of ME/CFS and a range of other conditions, including fibromyalgia (FMS), in which neuroinflammation may play a central role.
- Dig Deeper – Check out Marco’s Neuroinflammatory Series here
One difficulty in developing a model of ME/CFS in particular is the heterogeneity of symptoms and the range of putative triggers. The onset of FMS is often idiopathic.
Complex Regional Pain Syndrome

The pain in chronic regional pain syndrome (CRPS) is intense – even exceeding that found in fibromyalgia
Complex Regional Pain Syndrome 1 (CRPS) (previously known as Reflex Sympathetic Dystrophy or RSD) is a relatively rare but possibly under-diagnosed condition that has been recognized for some 150 years. (It was included by the eminent and influential 19th-century neurologist Charcot in the ‘hysteria minor‘ group of disorders.)
Some consider CRPS and fibromyalgia as close cousins, while others seem to want to put some clear blue water between the two based on the more intense pain experienced by CRPS patients (clocking in at a whopping 42 out of 50 on the McGill Pain Scale 2). Regardless, there’s extensive symptom overlap between the two disorders 3 and, by extension, with ME/CFS.
CRPS predominantly affects females (outnumbering men by a factor of four to one), and the most frequent age of onset is in the 40s. But it also occurs in children where it may be associated with Ehlers-Danlos Syndrome, mitochondrial disorders, or nerve entrapment. Most often CRPS develops following a relatively minor injury to a limb (even a needle stick!) associated with obvious nerve damage (CRPS II or causalgia) or without (CRPS I).
What follows is a level of pain that persists long after the injury (or surgery) that is out of proportion to the actual tissue damage and that progresses to a multi-organ systemic illness. No apparent biological basis for the pain has been found.
Reflex sympathetic dystrophy was abandoned as a name as it didn’t properly reflect the disease process, yet the ‘sympathetic’ part is accurate, as activation of the sympathetic nervous system plays a key role in the disorder.
Primary and Systemic Symptoms
The primary symptoms of CRPS are first seen in the affected limbs and include intense burning pain; ‘tropic’, autonomic, vascular sudomotor and endocrine changes affecting skin color and temperature, sweating, hair and nail growth with frequent neurogenic edema and various motor dysfunctions – fixed dystonia (fixed abnormal posture), myoclonic spasms, tremors, paresis (loss of voluntary movement), loss of strength and muscle atrophy.
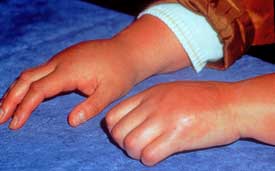
CRPS differs from FM and ME/CFS in that it produces swelling, color and skin changes, and sometimes even deformity in the afflicted limb. Other symptoms and findings, however, are quite similar.
Pain may also spread to other immediately adjacent areas, but can also turn up in areas far from the injury site.
Most intriguing are the systemic co-morbid symptoms that seem not only disproportionate to the initial injury but at first, glance seem completely unrelated and difficult to explain.
These include widespread hyperesthesia and allodynia, deep bone and joint pain, increased levels of pro-inflammatory cytokines suggesting systemic low-grade inflammation, cardiac symptoms (chest pain spreading to the arm, neck, and jaw, syncope, POTS, and increased heart rate and reduced heart rate variability reflecting generalized autonomic dysfunction) and gastrointestinal (including gastroparesis and reflux) and urological symptoms.
“The current CRPS study demonstrated increased heart rate and reduced heart rate variability. The pathologically reduced cardiac output and exaggerated increase in the total peripheral resistance during orthostatic stress point toward a dysfunction of the autonomic control of the cardiovascular system.”4
Both those statements could have come right out of a chronic fatigue researcher’s playbook.
Cognitive problems are also evident including problems with executive function, naming and memory :
“Significant neuropsychological deficits are present in 65% of patients, with many patients presenting with elements of a dysexecutive syndrome and some patients presenting with global cognitive impairment”5
Needless to say, as with other chronic illnesses – especially those involving pain – depression, anxiety, and symptoms of post-traumatic stress disorder – are common, as is fatigue:
“Almost all severely affected CRPS patients complain of lethargy, tiredness, and weakness.” 6
In one study 69% of patients described “overwhelming fatigue”.
Organic or Psychogenic?

The gender imbalance in CRPS and its many symptoms undoubtedly contributed to its designation as ‘hysteria minor’ despite its overt physical effects
Little wonder then that, since Charcot’s classification of CRPS as “hysteria minor”, these widespread systemic symptoms have often been interpreted and treated as psychogenic or given another euphemism like “medically unexplained”. Indeed, CRPS has also been called the “great imitator” due the overlap with or similarity to other disorders.
It holds that appellation in common with lupus, fibromyalgia and even sometimes chronic fatigue syndrome.
All now seems to be about to change. There is growing evidence, summarized in the following paper, that CRPS is a neuroinflammatory disorder and that it may be of autoimmune origin, with the result that the CRPS/RSD community is now discussing the likelihood of another name change.
Neuroinflammation, Neuroautoimmunity, and the Comorbidities of Complex Regional Pain Syndrome 7 – Mark S. Cooper and Vincent P. Clark
This intriguing paper sets out a model which, based on emerging evidence, attempts to explain the mechanisms by which a local peripheral injury can result, in a small minority of patients, in a widespread chronic multi-system condition. The key mechanism they propose is called spreading neuroinflammation.

As in ME/CFS and FM, a stressor, in this case often a small injury, can result in devastating changes for some. ‘Neuroinflammatory spreading’ could explain why…
Given that CRPS is a condition for which a neuroinflammatory process has been proposed and, unlike in ME/CFS or FMS, the initial trigger in many cases is unambiguous, CRPS may provide a more constrained environment in which to explore a potential neuroinflammatory model of other complex multi-system syndromes including ME/CFS and FMS.
It’s worth repeating that a) it often takes only a small injury to trigger this devastating disorder, and b) as occurs with post-viral ME/CFS or trauma induced fibromyalgia, only a minority of people develop it, and c) the symptoms of CRPS are quite distinct from the original trauma.
This is a complex paper, which I can only hope to skim over. For those of you interested, the full paper is well worth reading. I’ve attached a list of references and some related resources below.
Spreading Neuroinflammation and Neuroinflammatory Tracks
Experimental evidence supports the notion of spreading neuroinflammation where inflammation spreads from the initial injury site throughout the nervous system, either towards the periphery or to higher level regions.
Peripheral trauma or injury results in activated microglia at first order neurons. In a minority of patients, neuroinflammation migrates trans-synaptically to second order synapses at higher levels of the nervous system including the thalamus (an area of the brain involved in sensory perception, motor functions, sleep and alertness).

Inflammatory spreading through the nervous system appears to cause both central sensitivity and affect some of the same cognitive processes that are disturbed in ME/CFS…
In CRPS, it appears that a complex interaction of activated microglia, astroglia and NMDA (N-methyl-D-aspartate or glutamate) receptors in the spinal cord results in “wind-up” (a nerve process that exacerbates pain), central sensitization, loss of inhibitory tone, excessive sensory gain (pain signals are amplified) and reduced sensory gating.
First, an injury in the periphery (body) activates first-order nociceptive (pain-sensing) neurons to release the signaling cytokine CCL21, which then triggers secondary neuroinflammation in the thalamus, creating a ‘central generator of neuropathic pain’.
Neuroinflammation or neuroinflammatory tracks that spread through the nervous system alter neurotransmission and coding, causing higher-order functional changes in the nervous system including, perhaps, deficits in higher-level brain tasks such as executive functioning.
It’s important to note that these neuroinflammatory tracks may not necessarily result in easily detected structural changes to the nervous system (e.g. demyelination of nerves or gross structural changes to the brain). Functional or dynamic lesions that may also contribute to the pathology may be undetectable using neuroimaging technologies such as traditional MRI scans.
“Spreading neuroinflammation provides a non-psychogenic etiology to plausibly explain the progression and chronicity of certain disease states, as well as the migration of symptoms to different portions of the body. This mechanistic concept of spreading neuroinflammation within the CNS needs to be incorporated into differential diagnosis of neurological and neuropsychiatric disorders, as well as into the standard use of the Diagnostic and Statistical Manual for Mental Disorders (DSM). This would substantially advance the recognition and diagnosis of neuroinflammatory-mediated functional disorders within the biomedical community.”
Spreading neuroinflammation in ME/CFS and FMS?
Spreading neuroinflammation provides a mechanism by which a discrete peripheral injury can set up a chain of physiological and functional changes to the peripheral and central nervous system that results in a widespread, chronic systemic illness.
However, few ME/CFS patients report similar physical trauma around the time of onset, but a substantial proportion report onset following a viral illness. A neurotropic virus (one with an affinity for infecting neurons) is one possibility as long as the virus can cross the blood brain barrier. (One example is the Japanese Encephalitis virus, which causes neuroinflammation of the central nervous system, but no such similar virus has been found consistently in ME/CFS patients.)
Many report the onset of symptoms following common viral infections such as mononucleosis, and the Dubbo studies suggest that a small minority will develop ME/CFS following a variety of common infections, just as only a few develop CRPS following trauma.
Two speculative hypotheses suggest possible “hit and run” scenarios. In one, a sensory ganglia or paraganglia infection of the vagus nerve establishes a self-perpetuating activation of glial cells leading to ongoing sickness behavior 8. In the other, as demonstrated in an animal model, a common respiratory infection (a modified form of the H1N1 flu virus) can establish spreading neuroinflammation in glial cells contained in the olfactory bulb, which is anatomically close to key brain regions. The authors propose that repeated such encounters with common viruses might explain neurodegenerative conditions such as Alzheimer’s. 9
In contrast, estimates for precipitating factors in FMS vary from around 70% being idiopathic to up to 50% reporting onset following physical trauma.
Autoimmunity
Given that there is a clearly identified trigger, why then do only a small proportion of patients go on to develop CRPS? CRPS literature suggests that some people do appear to have a predisposition to developing conditions such as CRPS. (Given the history of the condition in the medical community it would be little surprise if these predispositions were presented as psychological or personality-based).
The gender bias (like ME/CFS and FMS), though, is suggestive of many autoimmune diseases, and some autoimmune diseases cause similar widespread systemic symptoms (Sjogren’s Syndrome), and autoantibodies to neurotransmitters can result in muscle pain, stiffness and dystonia (Stiff Person Syndrome).
Adrenergic receptor issues have shown up in both CRPS and chronic fatigue syndrome
An autoimmune contribution to CRPS now appears likely given recent findings that 90% of an adult CRPS cohort had autoantibodies (agonistic, therefore upregulating) to either the beta(2)-adrenergic receptor (β2AR) or the muscarinic acetylcholine receptor (M2R). Fifty percent of the cohort had autoantibodies to both. 10
The actions and distribution of these receptors closely match CRPS symptoms: β2AR or M2R receptors are found in the cerebellum, reticular formation, motor cortex, and thalamus in the brain, in the sympathetic and parasympathetic nervous system, the heart, the pyramidal motor pathway to skeletal muscles, in peripheral nerves, and in astrocytes and microglia.
Interestingly Alan Light’s team 11 recently found a threefold upregulation of gene expression for the adrenergic alpha2a receptor in ME/CFS patients following exercise. In addition, several studies have found autoantibodies to muscarinic acetylcholine receptors in ME/CFS 12, 13 and Sjogren’s Syndrome 14. Evidence for autoimmunity in FMS is scant but fibromyalgia does appear to occur co-morbidly in autoimmune diseases such as lupus, rheumatoid arthritis and autoimmune thyroid disease 15.
Peripheral injury can lead to a transient disruption of the blood/spinal cord and/or blood/brain barrier. In a CRPS patient with a peripheral nerve injury, circulating autoantibodies may infiltrate into the parenchyma (microglia and astrocytes) of the affected nerve causing a neuroautoimmune response as the immune system reacts to autoantibodies that bind to neuronal and glial cells. Autoimmune attack of peripheral nerves may then trigger spreading neuroinflammation in the spinal cord leading to the systemic co-morbid symptoms seen in CPRS.
Thus autoimmunity might sustain and exacerbate the spreading mechanism of neuroinflammation described above.
Small fiber neuropathy provides an intriguing potential link between FMS, CRPS and autoimmunity. Objective evidence of small fiber neuropathy has been found in FMS patients, most recently by the pain researcher Anne Louise Oaklander 16. Dr. Oaklander also studies CRPS patients and has found that those (Type I) patients, previously presumed not to have a nerve injury, actually have small fiber neuropathy 17. Could these previously undetected microscopic lesions be sufficient to trigger the infiltration of autoantibodies into the central nervous system either through the breakdown of the blood-brain barrier by pro-inflammatory cytokines or via peripheral glial cells?

Small fiber neuropathy has been found in CRPS/FM and ME/CFS patients. This image shows two highly degraded nerves; i.e., a neuropathy,
This model, therefore, provides a mechanism whereby peripheral inflammation (potentially exacerbated by autoantibodies to either the β2AR or M2R receptors) can propagate from the peripheral injury site to higher levels of the nervous system, resulting in hyperexcitability and eventually impacting higher cognitive functions (avoiding the blood-brain barrier issue by propagating via the parenchyma of the central nervous system).
The authors suggest that autoimmunity may be a predisposing factor and may develop in conjunction with a wide range of environmental or psychological stressors:
“The key concept here is that the development of autoimmunity to specific neuroautoantigens may be the initiating event for many cases of CRPS. Psychological stressors, physical trauma, infectious agents, and/or genetic susceptibility could all play a role in the breakdown of self-tolerance, and the onset of an autoimmune response. This set of etiological linkages fits well with documented clinical experience with CRPS (Mitchell 1872; Birklein et al. 2000). Psychological stressors and immunologic priming have been linked to the enhanced activation of microglia to nervous system injury (Frank et al. 2007; Hains et al. 2010).”
A note of caution, though: While the authors stress potential environmental triggers for autoimmunity, Professor Jonathan Edwards (who pioneered the use of B-cell depleting Rituximab in the treatment of autoimmune rheumatoid arthritis and who is advising on a UK trial of Rituximab in ME/CFS) has stated in discussions on Phoenix Rising that the development of autoimmunity is essentially a random event generated entirely by the immune system’s “accidental” production of antibodies that bind to “self” cells. From this perspective, environmental triggers may be innocent bystanders in the development of autoimmunity but, as with damage to peripheral nerves in CRPS, may provide the opportunity for the autoantibodies to enter the nervous system causing systemic disease.
CRPS Treatments
Current treatments for CRPS appear to be aimed at symptomatic relief and/or attempting to prevent further progression of the condition. Amongst these are a familiar collection of drug and non-drug therapies targeted at pain, neuropathic pain, and neuroinflammation.
Physiotherapy and psychotherapy are commonly used, while more invasive/radical treatments such as surgical sympathectomy and spinal cord stimulation have been tried
On- and off-label drugs include NSAIDS and corticosteroids which appear to be of little use once the condition has become chronic. Others (Neurontin, Lyrica, Cymbalta, Savella, Baclofen and Ketamine) work to either boost the inhibitory neurotransmitter GABA or block excitatory glutamate NMDA receptors – the balance between the two having been long implicated in chronic pain, movement disorders, and neuroinflammation generally. Ibudilast, the antibiotic Minocycline, and Low Dose Naltrexone may attenuate activated glia 18, which, as discussed above, are clearly implicated in neuroinflammation in CRPS. In contrast, long-term use of opioids may increase glial cell activation.
With immune activation implicated in CRPS, intravenous immunoglobulin (IVIG) treatment (antibodies extracted from healthy donors that may be helpful in some disorders of the immune system) has shown some efficacy in a small trial.
If autoimmunity does underlie CRPS, then previous treatment approaches are only addressing the symptoms and not the root cause. However, as the autoimmune findings are fairly recent (2011 in the case of the findings discussed above) B cell depletion therapy using agents such as Rituximab have not been tried in CRPS.
Reference 19 provides a more complete overview of treatment options and contraindications.
A (posthumous) Apology from M Charcot?
Has Charcot’s inclusion of CRPS within the hysteria minor disorders led to unnecessary skepticism and sub-optimal treatment from the medical profession over many decades? Possibly, but perhaps his students (including Freud) and the psychiatrist who claimed that Charcot knew “almost nothing about major psychiatric illness” 20 should have paid more attention to all of his writings before invoking ‘psychogenic’, ‘somatization’ or ‘conversion disorders’.

An era focused on psychological explanations passed over Charcot’s belief that undiagnosed lesions also played a role.
While Charcot may have dabbled in psychology and hypnotism, he was first and foremost a neurologist and to quote the main paper under discussion here:
In the 1880s, Charcot first hypothesized that hysteria was generated by non-structural lesions in the nervous system (Harris 2005). He postulated that these lesions were likely to be biochemical or physiological in character. In describing a case study, during a lecture on hysteria, Charcot (1885) stated: “We have here unquestionably one of those lesions which escape our present means of anatomical investigation, and which, for want of a better term, we designate dynamic or functional lesions.”
Charcot’s conception of disorders such as CRPS was clearly that they were due to physiological “lesions” that were not detectable by the then-current neurological means for investigation that depended on visible structural changes.
Or, in a nutshell, absence of evidence does not mean evidence of absence.
Perhaps now medical science and technology has advanced enough to detect microscopic structural lesions plus functional and temporal dysregulation of the nervous system of the type described here as “neuroinflammatory tracks” relegating “psychogenic” explanations of conditions such as CRPS and other “medically unexplained” symptoms to the past. 21
Could ME/CFS and FMS share a similar neuroinflammatory and possibly autoimmune etiology that, to date, has largely evaded investigators?
References and some Resources
1. Complex regional pain syndrome
Wikipedia http://en.wikipedia.org/wiki/Complex_regional_pain_syndrome
2. McGILL PAIN INDEX – WHERE IS CRPS PAIN RANKED?
American RSD Hope – http://www.rsdhope.org/mcgill-pain-index—where-is-crps-pain-ranked.html
3. Development of a Symptoms Questionnaire for Complex Regional Pain Syndrome and Potentially Related Illnesses: The Trauma Related Neuronal Dysfunction Symptoms Inventory
Susan Collins, MSc, Jacobus J. van Hilten, MD, PhD, Johan Marinus, PhD, Wouter W. Zuurmond, MD, PhD, Jaap J. de Lange, MD, PhD, Roberto S. Perez, PhD
4. Heart Rate Variability in Complex Regional Pain Syndrome during Rest and Mental and Orthostatic
Stress
Astrid J. Terkelsen, M.D., Ph.D, Henning Mølgaard, M.D., Ph.D., John Hansen, M.Sc.EE, Ph.D.,
Nanna B. Finnerup, M.D., Ph.D., Karsten Krøner, M.D., Troels S. Jensen, M.D., Ph.D.
http://www.rsds.org/2/library/article_archive/pop/Terkelsen_Anesthesiology_2012.pdf
5. Neuropsychological deficits associated with Complex Regional Pain Syndrome
http://www.rsds.org/pdfsall/Libon_Neuropsychol_2010.pdf
6. Systemic Complications of Complex Regional Pain Syndrome
Robert J Swartzman
7. Neuroinflammation, Neuroautoimmunity, and the Co-Morbidities of Complex Regional Pain Syndrome
Mark S. Cooper and Vincent P. Clark
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3661922/
8. Chronic fatigue syndrome from vagus nerve infection: a psychoneuroimmunological hypothesis.
VanElzakker MB.
http://www.ncbi.nlm.nih.gov/pubmed/23790471
9. Neuroinflammation Resulting from Covert Brain Invasion by Common Viruses—a Potential Role in Local and Global Neurodegeneration
Jeannine A. Majde, Ph.D.
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC2897933/
10. Autoimmunity against the β2 adrenergic receptor and muscarinic-2 receptor in complex regional pain syndrome.
Kohr D, Singh P, Tschernatsch M, Kaps M, Pouokam E, Diener M, Kummer W, Birklein F, Vincent A, Goebel A, Wallukat G, Blaes F.
http://www.ncbi.nlm.nih.gov/pubmed/21816540
11. Post-exertion malaise in chronic fatigue syndrome: symptoms and gene expression
Jacob D. Meyera, Alan R. Light, Sanjay K. Shuklad, Derek Clevidence, Steven Yale, Aaron J. Stegner & Dane B. Cook
http://www.tandfonline.com/doi/abs/10.1080/21641846.2013.838444#.UneiYvkqiSo
12. Autoantibodies against muscarinic cholinergic receptor in chronic fatigue syndrome.
Tanaka S, Kuratsune H, Hidaka Y, Hakariya Y, Tatsumi KI, Takano T, Kanakura Y, Amino N.
http://www.ncbi.nlm.nih.gov/pubmed/12851722
13. Reduction of [11C](+)3-MPB Binding in Brain of Chronic Fatigue Syndrome with Serum Autoantibody against Muscarinic Cholinergic Receptor
Shigeyuki Yamamoto, Yasuomi Ouchi, Daisaku Nakatsuka, Tsuyoshi Tahara, Kei Mizuno, Seiki Tajima, Hirotaka Onoe, Etsuji Yoshikawa, Hideo Tsukada, Masao Iwase, Kouzi Yamaguti, Hirohiko Kuratsune, Yasuyoshi Watanabe
http://www.plosone.org/article/info%3Adoi/10.1371/journal.pone.0051515
14. Clinical associations of autoantibodies to human muscarinic acetylcholine receptor 3213–228 in primary Sjögren’s syndrome
L. Kovács, I. Marczinovits, A. György, G. K. Tóth , L. Dorgai, J. Pál , J. Molnár and G. Pokorny
http://rheumatology.oxfordjournals.org/content/44/8/1021.full
15. Fibromyalgia and Autoimmune Disease : The Pain Behind Autoimmunity
Dan Buskila, MD, Piercarlo Sarzi-Puttini, MD.
http://www.ima.org.il/FilesUpload/IMAJ/0/42/21053.pdf
16. Objective evidence that small-fiber polyneuropathy underlies some illnesses currently labeled as fibromyalgia.
Oaklander AL, Herzog ZD, Downs HM, Klein MM.
http://www.ncbi.nlm.nih.gov/pubmed/23748113
17. Is reflex sympathetic dystrophy/complex regional pain syndrome type I a small-fiber neuropathy?
Oaklander AL, Fields HL.
http://www.ncbi.nlm.nih.gov/pubmed/19557864
18. Treatment of Complex Regional Pain Syndrome (CRPS) Using Low Dose Naltrexone (LDN)
Pradeep Chopra and Mark S. Cooper
http://www.ncbi.nlm.nih.gov/pmc/articles/PMC3661907/
19. Complex Regional Pain Syndrome or Reflex Sympathetic Dystrophy
Updates on Treatment
Pradeep Chopra MD
http://www.rsds.org/education/CRPS%20Updates%20on%20treatment%20CA%20-%20Chopra.pdf
20. Jean-Martin Charcot
Wikipedia
http://en.wikipedia.org/wiki/Jean-Martin_Charcot
21. Rethinking the Psychogenic Model of Complex Regional Pain Syndrome.
Renee J Hill, Pradeep Chopra, Toni Richard
http://www.rsds.org/pdfsall/ReneeHill-Broatch.pdf
Resources
Complex regional pain syndrome
NHS Choices
http://www.nhs.uk/Conditions/Complex-Regional-Pain-Syndrome/Pages/Introduction.aspx
Complex Regional Pain Syndrome Fact Sheet
National Institute of Neurological Disorders and Stroke
Reflex Sympathetic Dystrophy Syndrome Association – http://www.rsds.org/2/what_is_rsd_crps/
Complex Regional Pain Syndrome – Practical Pain Management.com
http://www.practicalpainmanagement.com/complex-regional-pain-syndrome-systemic-complications



I would take exception to Dr Edwards “theory” that autoimmunity is a random event. There are numerous “autoimmune” diseases in veterinary medicine that are clearly triggered by outside, particularly infectious agents. One in particular that is well studied and documented is autoimmune hemolytic anemia triggered by Ehrlichia organisms. Even once the organism is gone the disease persists, many others exist.
Greg
Yes, I have Ehrlichia as one of my co-infections and for sure it affects the immune system or doesn’t in my case. It goes undetected by the immune system and creates havoc in my body along with the one that is even bigger – Protomyxzoa Rheumatica, which is even harder to treat. Not sure how you treat it with animals but the key for me is to break down the biofilms that it hides in and activate the immune system to detect it. Once the biofilms are broken down with enzymes, then I take antibiotics and anti-malarial herbs to go for the pathogens. Seems to be making a difference. But, as there are cycles that these things go through – it’s not an easy fix or something that may ever be completely eradicated. Something I will have to work with for life.
Issie
Thanks Greg
Prof Edwards was pretty adamant that environmental triggers are usually a red herring or maybe I over-interpreted what I was saying. I’m certainly not qualified to argue the point.
Environmental triggers of autoimmunity might appear to fit better with viral onset theories of ME/CFS. I’m leaning more to environment and autoimmunity being co-factors.
I agree. I was perfectly healthy at 19 years. I was attending Ohio University and very active when the campus was hit with Rubella( 3 day measles) I was in the Clinic and at one time had a fever of 104. I was released after 3 days-still had spots-and the pain I experienced when the fever was so high never left. I’ve been battling this my whole life. In 1964 there was no diagnosis. I am 78 and now the pain has become very intense. I was finally diagnosed by the Cleveland Clinic with Sjogrens, ME/CFS in the 90s. I was athletic and fine I got Rubella- afterwards I was in Chronic Pain and Fatigue. To me it’s always been obvious. Rubella was the illness that if Pregnant Women got it their Babies were severely affected- deaf, deformed etc. If it could do that why wouldn’t it effect a 19 year old girl whose body was still changing.
Thank You
Kathleen Stringer
Well Done Marco! You were not kidding when you said I’d like it. There are so many levels of relevance here to what many of us are dealing with.
Having EDS and FMS myself and having gone through a whole lot of trauma with my body (surgeries and injuries) and being in pain a good bit of the time – this is especially interesting. Also, having hypogammaglobulinemia and known autoimmune issues (vitiligo, alopecia) and now the association with the immune system to POTS —this seems very relevant to me. Working on my immune system has been my best treatment to date.
I also have found Tramadol (pain med) and Bentyl (muscle relaxer) to be one of my best POTS and EDS treatments too. Those meds work on so many levels of what is discussed here – GABA and opiate receptors, serotonin and dopamine. And the muscle relaxers work on GABA also..
The other modifiers that I use to address my immune system are doxycycoline and GastroCrom and, of course, my DIET.
When I have my CRP levels checked they are really high and this is despite herbals to calm down inflammation. I asked my doc how this could be and he said that I’m still not addressing whatever is causing the inflammation. We need to find another cause and another solution. I wonder how bad it would be without the help that I have with the herbals. How high would those inflammation levels be?
One alternative to IVIG is colostrum and this is something that I use and find helpful. It is also very calming to the nervous system. (I use the Immune Formula of Symbiotics Colostrum.)
I’ve never really thought to put the label of Regional Pain Syndrome on myself – but, this could explain a lot. Interesting connection here and it makes sense that autoimmune and inflammation are our keys. Reinforces the way I think about things as to our “cause”.
Thanks for the write up Marco.
Issie
Thanks Issie
I do feel that things are starting to come together pretty rapidly for a range of conditions perhaps driven in part by new technologies. There also seem to be more researchers prepared to think outside of traditional boxes – or maybe I’m just naturally drawn to that kind of paper.
If you get the chance – do read the main paper I referenced – I was only able to give the barest outline.
Ridiculous though in this day and age that CRPS researchers still feel the need to rebut psychogenic explanations.
Issie, I;m curious as to what type of enzymes you are taking to help break down the biofilm. For the past week I’ve worked on lowering my daily intake of fats, 5 days with low fat consumption and 2 with moderate. On all of the low fat consumption days I had no night sweats but with the 2 days of moderate consumption I had mild night sweats. It’s looking more and more like I too might have a Protomxyzoa Rheumatica infection or at least something similar.
I use Lumbrokinase at least two hours before taking either the antibiotic or anti-malarial herb and then two hours after those, I take probiotics. First you breakdown the bio-films, go in for the attack and then support with good bacteria. You don’t want a yeast problem on top of it all.
I have also found that a low-fat diet makes a difference with me too. The other thing that is important when you are dealing with the immune system is to support it with what we know. One of the first things that some of the known autoimmune disorders (MS) are told to avoid is dairy. And then the second thing is gluten (wheat, rye, barley). I find this really makes a difference for me. In order to stick with anything, we have to educate ourselves on the why. There has to be a good reason for the decisions we make. Then we are more likely to stick with it. Watch the videos that are on line for this lifestyle. (Dr. McDougal – Starch Solution, Forks over Knives, Engine 2 —all of these are a good place to start.) Make sure that you eat as Whole Foods as you can – not refined. You want to do it smart and have a variety of veggies, fruits and whole grains. Also, make sure you get enough plant proteins. This has really helped my intestional health – I seldom have problems any more.
One other thing you might think about in regards to the sweats – pay attention to what type of foods you are eating on those days. Could you be eating more histamine forming foods? Could there be a possible mast cell problem? I found keeping a food journal helped me to analyze things closer in my first few months. It helped me to identify triggers that were very enlightening.
Keep us posted on how you do.
Issie
Marco, thank you for posting this article. I found it to be very pertinent to something that happened to me. I fell on concrete and landed on my elbow shattering some bones. I had surgery to remove the shattered bones as nothing could be done with pins and so forth.
Within a week or two, I developed RSD, as it was called then, in my hand on the same side as the surgery. My hand became quite swollen, turned purple and had sort of a sheen to it. Everyone who saw it couldn’t help but exclaim over it. It was very grotesque.
Just one month before, I saw an ME/CFS specialist who diagnosed me with Fibro and CFS. Up to this point, I did not have any cognitive difficulties. After the broken elbow, surgery and RSD, they began to develop.
The surgeon took a wait and see approach with the RSD. Fortunately, it resolved on its own. I believe the neurontin, which I was taking at the time, knocked the edge off the severe pain.
Once again, thank you for the explanation to something that has puzzled me for a long time.
Thanks for passing that on. Perhaps Neurontin’s ability to cut glutamate production helped stop the immune activatiion associated with RSD.
How interesting that that ‘package’ of symptoms – pain, cognitive difficulties, fatigue etc – the hallmarks of ME/CFS/FM – began to develop at once. Some researchers refer to that at the ‘fibromyalgianess of disease’ and it appears to happen in a subset of every pain producing disorder.
Glad you got out of the RSD – what a difficult disease that is….
Hi Polly
Thanks for that. What an intriguing (and painful) story.
So, if I picked this up correctly, you had already been given FMS/CFS diagnoses before the accident and subsequently developed RSD/CRPS including new cognitive problems?
If CRPS has an autoimmune component does this suggest that autoantibodies were already there before the accident and may have contributed to your FMS/CFS?
Sounds like a lucky escape with the Neurontin. Was this prescribed following the injury or were you already taking it?
One of the CRPS specialists recommends that CRPS patients should take similar meds in advance of even minor surgical procedures to head off any chance of it getting worse.
Thanks again and I’m glad it seems to have provided some answers.
Very interesting Marco,
Was that Lyrica you were referring to? I remember hearing that taking it pre-surgery could help cut down on the risk of having chronic pain later.
Hi Cort
That’s from reference 19 :
http://www.rsds.org/education/CRPS%20Updates%20on%20treatment%20CA%20-%20Chopra.pdf
Pre operative precautions
avoid surgery unless you have to (mmm -seems redundant):
start gabapentin or pregabalin 2 weeks before;
Minocycline 1 day before and for two weeks following;
Vitamin C 500mg one daily. Start before surgery and continue for 45 days after surgery.
He then goes on to discuss intra and post operative precautions.
It makes me wonder what the risk of developing CRPS is in FM or ME’/CFS and whether they should take these precautions as well.
Why not at least take Lyrica pre-surgery? Numerous studies have found Lyrica effectively reduces pain after surgery and some have found that taking it pre-surgery is helpful….and reduces opioid consumption after surgery.
Surg Endosc. 2013 Jul;27(7):2504-11. doi: 10.1007/s00464-012-2769-3. Epub 2013 Jan 24.
Effect of pre-emptive pregabalin on pain intensity and postoperative morphine consumption after laparoscopic cholecystectomy.
Sarakatsianou C, Theodorou E, Georgopoulou S, Stamatiou G, Tzovaras G.
CONCLUSIONS:
Administration of 600 mg pregabalin per os, divided in two preoperative doses, significantly reduces postoperative pain as well as opioid consumption in patients undergoing laparoscopic cholecystectomy, at the cost of increased incidence of dizziness.
I find this
“Psychological stressors, physical trauma, infectious agents, and/or genetic susceptibility could all play a role in the breakdown of self-tolerance, and the onset of an autoimmune response.”
so interesting with regard to the onset of autoimmune disorders.
Check out this sentence from an abstract on autoimmunity and CRPS
“These findings suggest that neuroinflammatory lesions, as well as their associated functional consequences, should be evaluated during the differential diagnosis of non-dermatomal pain presentations, atypical movement disorders, as well as other “medically unexplained symptoms”, which are often attributed to psychogenic illness.”
And another which suggests the autoimmunity present in CRPS is unusual
“We propose that CRPS constitutes a prototype of a new kind of autoimmunity, which we term ‘IRAM’ (injury-triggered, regionally-restricted autoantibody-mediated autoimmune disorder with minimally-destructive course). Understanding autoimmune contribution to CRPS should allow the exploration of novel treatment modalities in the future.”
There’s alot to learn regarding autoimmunity CRPS has been recognized since the 40’s but it wasn’t until 2004 I think it was that research into autoimmunity began, and only in the last couple of years has it become something of a major topic. Autoimmunity comes in different forms and can obviously take time to pin down…..We’ll see what happens in ME/CFS….
Hi everyone, I also went from being fine to being sick. I started with flu like symptoms and then pain in my R arm with swelling and levitigo reticulitis. Many Dr’s visits and ER visits later I was diagnosed with CRPS and then stared developing Fibromyalgia pain and working at a very stress full job I started feeling so fatigued. It was like I could feel the gravity. I worked sick for 4 years until I literally collapsed and developed an ulcer from the Tramadol. I met a Doctor named Dr. Rey at UM and she was able to draw special labs and found that I also had CFIDS. I am on disability, it is very sad because I am young and was at the top of my career and had just finished a Master’s degree in Nursing. Now the RSD is more prominent in my R leg. The R hip and R knee were both MRI’d and I need to have a knee replacement. I walk with a cane and my R side is weaker than the L. Thank you for this article because it describes me and what I have been through. I need validation. We all do with invisible diseases. Ivette
Thanks for so evocatively describing what CRPS is like; good luck Ivette!
Most of this is a little above me.. But I do have CRPS and now fibro. I wish I had of know there was even the slightest chance of getting CRPS or any info on what could go wrong.. I had surgery for carpel tunnel.. It took about 45 mins to release the nerve.. I didn’t know that the swelling and pain was CRPS .. I was given no meds for pain even after diagnosis. I received physio for months my hand is still not right but it is better..
Any little break through that Drs can make with these diseases is greatly appreciated
Thank you
Marco, your forensic biochemical detailing of the processes is impressive and we are fortunate to have someone who is willing to put in that much research effort (and has the brains to do so, of course). So far you have covered the nervous system and immune systems but these illnesses are neuro-endocrine-immune and I look forward to you revealing the same forensic detail of the role the endocrine system plays in these processes. Maybe the answer can be found there! And why is it that trigger points are never mentioned when they can cause pain up to screaming point and have many perpetuating factors, including hormone imbalance and viruses. They also develop from trauma, even the most minor trauma if the other perpetuating factors are many.
Here is a list of the authors of past and present day who are the primary source for trigger points and myofascial pain therapy. Myofascial tissue disease is at the core of all complex pain syndromes ie RSD/CRPS
>Intramuscular Stimulation using the techniques of C. Chan Gunn, MD.
>Trigger Point Injections using the techniques of Janet G, Travell, MD, David Simmons, MD and Edward Rachlin, MD.
>Ligament and tendon relaxation techniques of George Stuart Hackett, MD.
>CraigPENS as per William F Craig, M.D.
>Myofascial Release by Gokavi, Cynthia N. Gokavi, MBBS.
>The Trigger Point Therapy Workbook: Your Self-Treatment Guide for Pain Relief, Second Edition by Clair Davies, Amber Davies and David G. Simons (Aug 1, 2004)
>Fibromyalgia and Chronic Myofascial Pain: A Survival Manual (2nd Edition) by Devin J. Starlanyl and Mary Ellen Copeland (Jun 30, 2001)
>Advanced Soft Tissue Techniques as per Leon Chaitow, ND, DO
>Medical Acupuncture as per French Energetic protocols of Joseph Helms, MD.
None of these guy were mentioned?
Many thanks for the information Dr Rodrigues
As you may have guessed this blog was intended to highlight one particular theory of CRPS that might have relevance to FMS and CFS rather than being a comprehensive overview of the condition.
So again thank you for filling in some of the missing details.